Bulk Transcriptomic Deconvolution Profiles Survival Linked Immune,Stromal, and Unclassified Malignant Like Cell States in ccRCC
This study integrates bulk RNA-seq, single-cell atlases, and cell–cell communication analysis to show that the large “UNKNOWN” fraction in ccRCC deconvolution represents misclassified malignant tumor cells, highlighting key limitations of reference-based methods and the need for tumor-inclusive models.
Our motivation
Our vision is to improve the biological interpretability of bulk transcriptomic deconvolution in clear cell renal cell carcinoma (ccRCC) by resolving the identity of large, unexplained cell fractions that emerge when tumor intrinsic states are absent from reference matrices. By integrating bulk RNA-seq with single-cell atlases, we aim to bridge methodological gaps between population-level inference and cellular-level ground truth, ensuring that malignant transcriptional programs are accurately represented rather than obscured as unclassified signal
Our vision
Reference-based deconvolution tools such as CIBERSORTx frequently produce substantial “UNKNOWN” fractions in ccRCC tumors when normal tissue atlases are used, raising concerns about biological misclassification rather than technical noise. Given the strong hypoxia-driven and metabolically reprogrammed nature of ccRCC tumor cells, we were motivated to test whether these UNKNOWN signals reflect malignant transcriptional states that collapse when tumor-specific signatures are missing, potentially misleading downstream survival and microenvironmental interpretations
Output
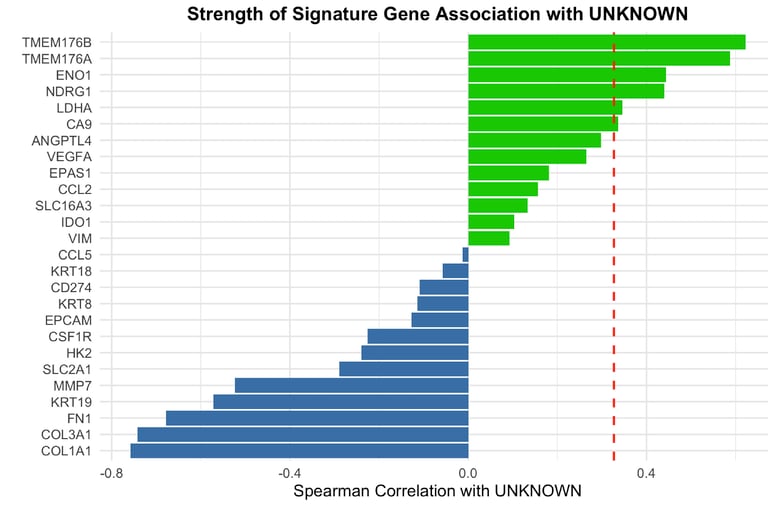
Our analysis demonstrates that the UNKNOWN fraction in TCGA-KIRC bulk RNA-seq data represents misclassified malignant tumor cells rather than immune or stromal populations. This conclusion is supported by strong correlations with canonical malignant markers, consistent enrichment of hypoxia-associated programs, and dominant tumor-driven communication patterns. Importantly, while UNKNOWN abundance varies across survival quartiles, it does not track with poor prognosis, highlighting that its biological meaning is tumor intrinsic rather than prognostically immune-driven
Tools Used
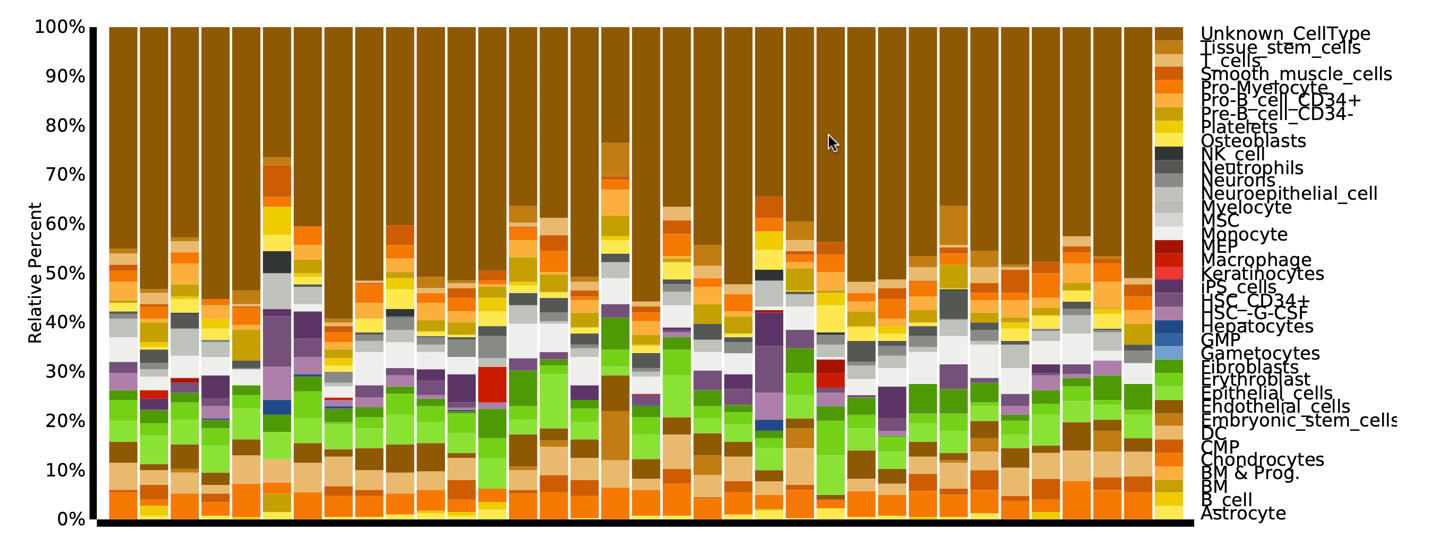
This study leveraged CIBERSORTx for bulk transcriptomic deconvolution of TCGA-KIRC samples using an HPCA reference, enabling systematic quantification of immune, stromal, and unclassified fractions. Single-cell RNA-seq integration from TISCH datasets provided tumor-inclusive cellular resolution, while CellChat was used to model ligand-receptor communication networks and signaling dominance across cell states. Together, these tools enabled cross-modal validation of malignant identity from expression, correlation, and intercellular signaling perspectives.
Single-cell analysis
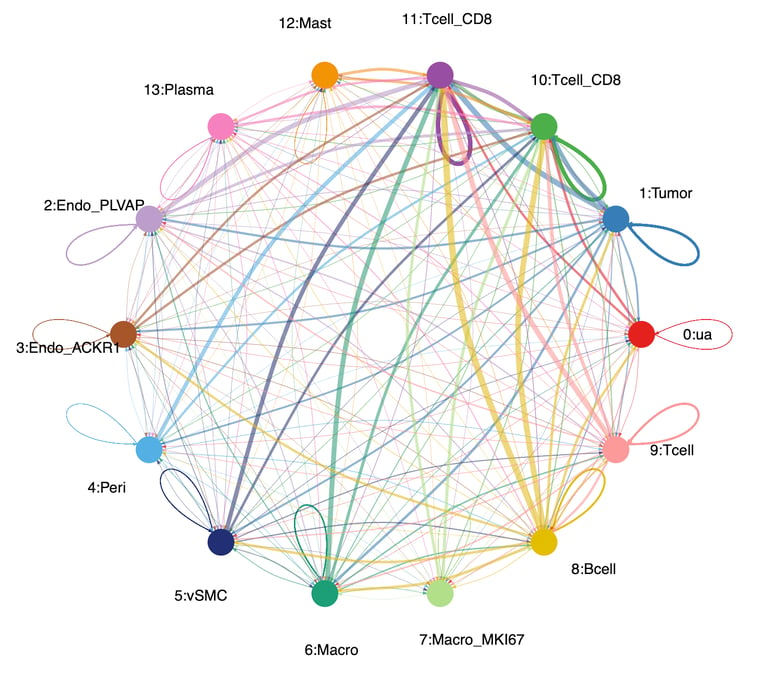
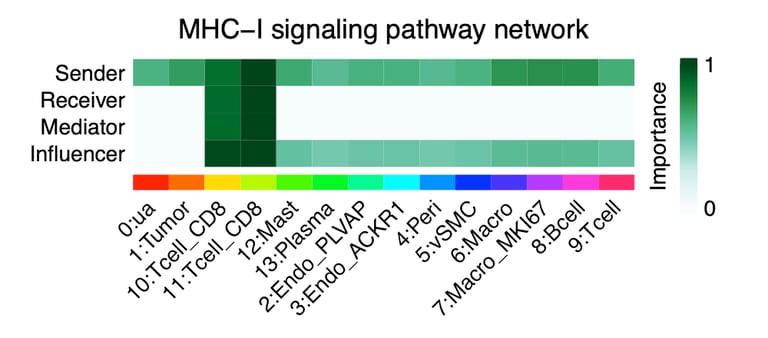
To validate the cellular identity of the UNKNOWN fraction, we integrated two independent ccRCC single-cell RNA-seq datasets and projected malignant marker expression and composite tumor signatures onto UMAP embeddings. Malignant markers such as CA9, TMEM176A/B, ENO1, and NDRG1 localized exclusively to tumor clusters, with minimal expression in immune or stromal compartments. Cell-cell communication analysis further revealed malignant cells as dominant senders and mediators of MHC-I and tumor-immune signaling, confirming that UNKNOWN reflects active malignant biology.
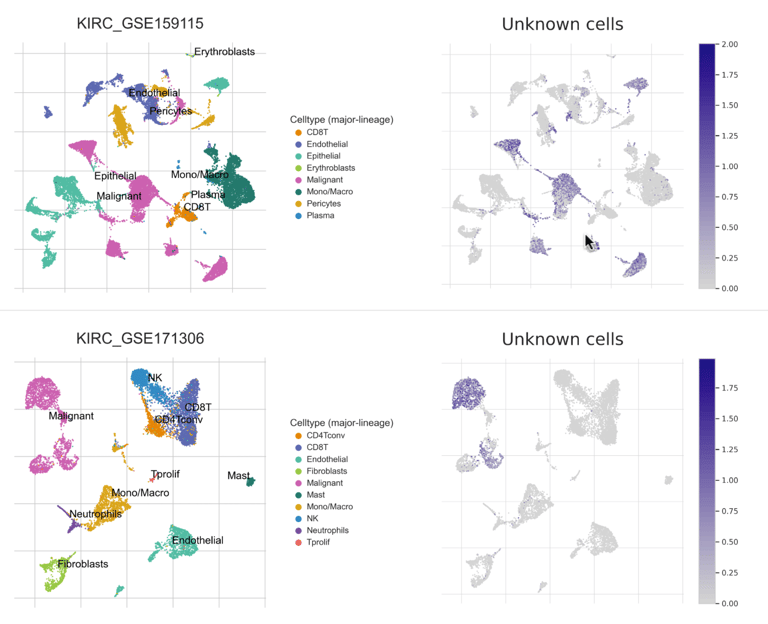
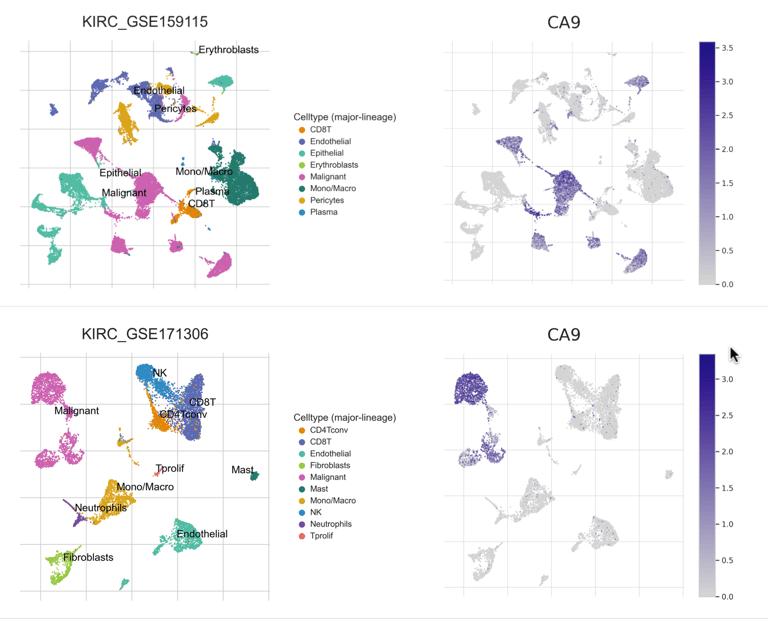
CellChat Analysis
CellChat analysis was used to characterize intercellular communication patterns within ccRCC single-cell datasets by modeling ligand–receptor interactions across malignant, immune, and stromal populations. The analysis revealed that malignant tumor cells corresponding to the bulk “UNKNOWN” fraction act as dominant signal senders, receivers, and mediators, particularly through MHC-I, VEGF, CD70, and MIF signaling pathways. These communication networks showed strong tumor–CD8 T cell interactions and minimal stromal contribution, supporting the conclusion that the UNKNOWN fraction reflects active, tumor-intrinsic signaling rather than microenvironmental cell types.